機械学習で物性予測!
こんにちは!CheMLです.
前回はpythonのscipyライブラリを使ったピークフィッテイングについて紹介しました.機械学習手法を紹介するといいつつ初っ端から全然機械学習関係ない記事でした(汗).
今回はいよいよ機械学習(Tensorflow)を使った化学データの解析です.各種分子の構造式とHOMOエネルギーとの相関をニューラルネットワークで学習して,分子構造からHOMOエネルギーを推定しよう!という作戦です.
結論から申し上げますと,トレーニングデータ・テストデータともに良好なフィッティングができまして,分子構造からかなり正確にHOMOエネルギーを予測することができました.
Tensor flowとは...
まず初めに,今回機械学習のエンジンとして用いたTensorflowについて簡単に紹介します.
pythonでの機械学習では,十中八九次の2種類の機械学習ライブラリが使われます.
- scikit-learn
- Tensor flow
TensorflowはGoogleが開発する機械学習ライブラリで,ニューラルネットワークを中心に様々な学習プログラムを簡単に実装できます.また,GPU計算を簡単に導入できることも特徴の一つです.個人的には人工ニューラルネットワーク(ANN: artificial neural network)に関してはTensor flowの方が使い勝手がいいと思いますので今回はTensorflowを使っていきます.
QMデータセットについて
今回入力に使用するデータセットはQM9というデータベースです.(QM9データセットはこちらからダウンロードできます.)
QM9は,GDB-17というデータベースからC, N, O, Fが9個以下の分子(133,885種)を抽出し,最適化構造と各種物性値をまとめたものです.(GDB-17データセットはこちらからダウンロードできます.)QM9の他にも異なる条件で分子を集めたQM7やQM8などのデータベースが公開されています.
QM9には各分子のB3LYP/6-31G(2df,p)レベルでの最適化構造がSDF形式で,各種物性値がCSV形式で含まれています.そのため,構造と物性値の相関関係を調査するのに適したデータセットです.
ピロールのHOMOエネルギーを予測しよう!
それでは具体例としてピロールのHOMOエネルギーをニューラルネットワークで予測してみましょう.今回は時短のため,QM9データセットの中からピロールと類似した化学構造を持つ化合物を抽出して19,273分子を使いました.このうち20%をテストデータ,80%をトレーニングデータにしています.
プログラムの流れは以下の通りです.
- データの読み込み
- 分子構造でフィルタリング
- 外れ値を除去(平均値から標準偏差の3倍以上離れたサンプルは除去)
- 分子構造をフィンガープリントに変換(今回はMorganフィンガープリントを使用しました)
- フィンガープリントをxに,HOMOエネルギーをyに格納
- フィンガープリントをnumpy array型に変換
- 変数のオートスケーリング
- ニューラルネットワークによる学習
- トレーニングデータとテストデータのプロット
- 学習したモデルからピロールのHOMOエネルギーの推定
やることが多いので結構大変です...ちなみに今回学習に使用したニューラルネットワークの構成は以下の通りです.
________________________________________________________________
Layer (type) Output Shape Param #
=======================================
dense (Dense) (None, 100) 409700
________________________________________________________________
dense_1 (Dense) (None, 10) 1010
________________________________________________________________
dense_2 (Dense) (None, 1) 11
=======================================
インプットレイヤーを100個のニューロンで構成し,10個のニューロンでできた中間層を挿入しています.なお,すべての層にL2ノルム正則化を施してオーバーフィッティングを防いでいます.
最適化アルゴリズムはAdamで,学習率を5e-4に指定しています.最大epochは150回ですが,early_stopオプションを付けているので,損失関数の変化が小さくなると自動的に学習を終了します.(実際には118epochで終了しました)
ちなみに,プログラムが正常に進んでいるか確認するためにところどころに経過時間を表示するコードを挟んでいます.
長くなりましたが,以下がソースコードです.
gist14989bb644dc328f6001380d638cd0d0
学習と推定の結果
学習の結果,トレーニングデータの決定係数R2値は0.8857,テストデータは0.8316でした!なかなか高い相関だと思います.以下が実行結果のグラフです.

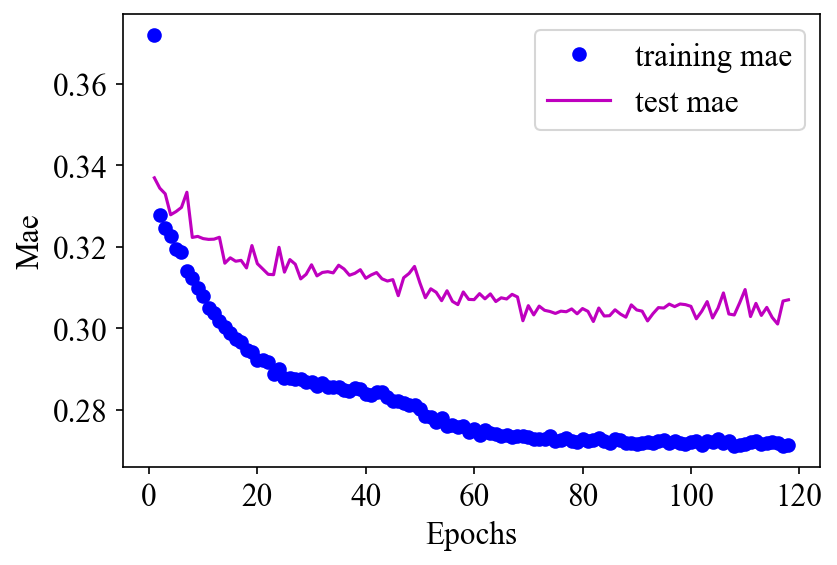

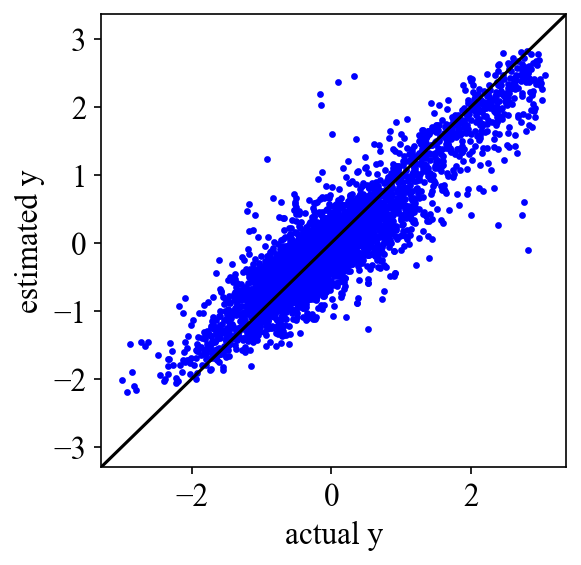
学習されたモデルを使ったピロールのHOMOエネルギーの推定結果は-0.211 hartreeでした!実際のピロール(ID: gdb_50)のHOMOエネルギーは-0.203 hartreeなので誤差は3.79%です.なかなかよく一致しているのではないでしょうか.
まとめ
以上のように,ニューラルネットワークを使うと物性値の予測が簡単に行えるようになります.このような経験的な手法で物性値を推定する方法にはClogPやJoback methodが有名です.ClogPはlogP(溶解の分配係数)を,Jobackは熱力学諸量を経験的に推定するための手法です.どちらも化学の現場ではよくつかわれています.
ニューラルネットワークや最適化,フィンガープリントに関する詳しい話はまた改めて記事にする予定です.
ご意見・ご感想はコメント欄,またはこちらまで.フォローしていただけるとやる気が出ます!
実行環境
Spyder(anaconda3)
python 3.7.9
windows10
*2020年9月現在では最新版のpython3.8.5に対応していないライブラリも多いため,計算化学ユースにおいてはpython3.7の使用をお勧めします.
Pythonでピークフィッテイング
こんにちは!CheMLです.
今回はPythonを使ったピークフィッテイングについての紹介です.
ピークフィッテイングができるソフトやプログラムはたくさんありますが,もちろんpythonでも可能です.Excelのソルバーでも簡単に実装できますが,速度や使いやすさの観点ではpythonの方が断然おすすめです!それでは早速フィッティングを行っていきましょう.
Gaussianフィッティング

今回はXPS(X線光電子分光)のスペクトル(酸素1s軌道)を用意しました.scipy(※1)のoptimizeパッケージに入っているcurve_fit()という関数を使ってGaussianフィッティングを行います.使い方は簡単です.curve_fitの中身に必要な情報を引数として与えるだけでフィッティングができちゃいます.
curve_fit(関数, 入力データx, 入力データy, [初期値])
ちなみにcurve_fit関数の最適化アルゴリズムにはlevenberg-marquardt法というものが使われています.これはGauss-Newton法と最急降下法を組み合わせた手法で,現在幅広く用いられている最適化の手法です.levenberg-marquardt法については今後詳しく解説する予定です.
curve_fitは戻り値としてフィッティングされたパラメータと係数の共分散行列を返します.そのため,空の変数はp_optとp_covの2つ用意しています.あとは結果のプロットと決定係数の計算,そして結果の出力(csv)です.決定係数の計算は手動でやっていますが面倒な方はpandasのcov()メソッドやscikit-learnのmetrics.r2_score()を使ってもいいと思います.
以下がソースコードです.
gist433a98bfdc7f57973afc3295a1f69c39
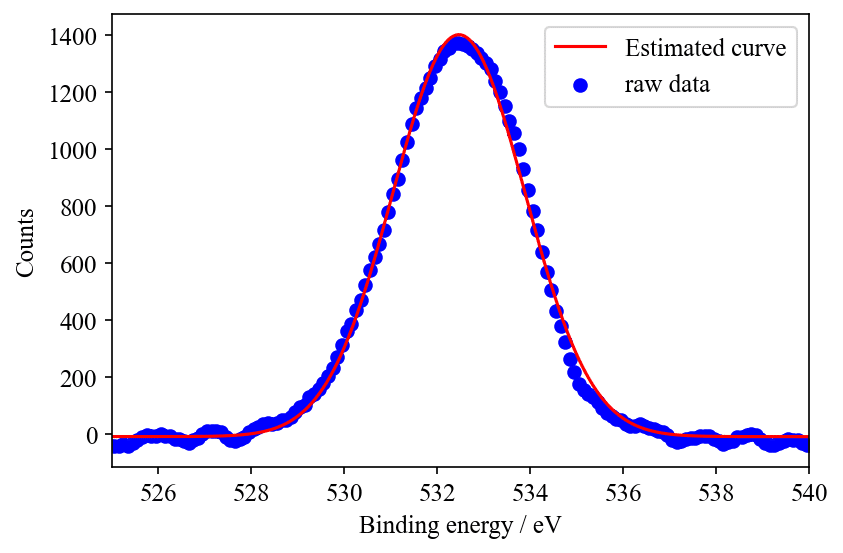
以下が実行結果です.savefig()で得られたpng画像を載せました.きれいにフィッテイングできていますね.
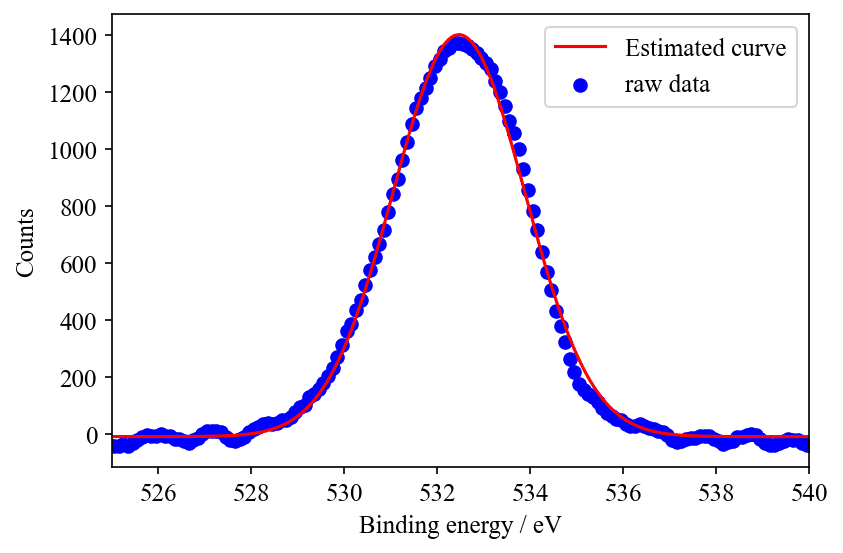
(ちなみに高解像度な画像を得るにはオプションにdpi=300を追加するといいです.300くらいで論文・レポートには十分だと思います.Transparent=Trueとすることで背景が透明になります.bbox_inches="tight"は画像サイズをグラフサイズに合わせるためのオプションです.)
各種パラメーターは以下にように表示されました。決定係数は0.9962と良好にフィッテイングできていることが分かります。
gista8299f8fa1a8d33d2ff9b6a09fdd4a5f
擬Voigtフィッティング
以上ではpythonでGaussianフィッティングする方法を紹介しました.しかし実際にはXPSのスペクトルはハイゼンベルグの不確定性幅(寿命に関する線幅,τは寿命,ℏはディラック定数)
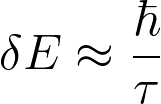
を持つためGaussianとLorentzianを畳み込んだVoigt関数を使用すべきですよね.(*Voigt関数は収束性は悪いため畳み込みの代わりにGaussianとLorentzianを足し合わせた擬Voigt関数を用いることが多いです.)
上記のソースコードを以下のように改造すれば擬Voigt関数でのフィッティングが行えます.
gist2550909c75de0eee8eb988d8674e579f
以下が実行結果です.Gaussianフィッティングとほとんど差異がありません.
gist9dde479aa46f0c2a03365036129ba2d6
Gauss関数の割合が1.00にフィッティングされていて,全くLorentzianの寄与がないことが分かります.なおGaussianの割合は使用した装置によってかなり異なるので注意が必要です.
まとめ
以上,今回はscipyのcurve_fitを用いたピークフィッテイングでした!
化学実験のデータ解析では頻繁に使える手法ですので,ぜひ有効活用してみてください.
ご意見・ご感想はコメント欄まで.フォローしていただけるとやる気が出ます!
実行環境
Spyder(anaconda3)
python 3.7.9
windows10
*2023年3月現在では最新版のpython3.8.5に対応していないライブラリも多いため,計算化学ユースにおいてはpython3.7の使用をお勧めします。
※1 Scipy: 科学技術計算用pythonライブラリ(今回のようなフィッティングだけでなくスムージングや関数の微分積分など幅広い演算が可能です。本noteでも多用します。)
★筆者のつぶやき★
「XPSスペクトル」はスペクトルを2回言ってるので「XPスペクトル」が正しいけど違和感ありますね...笑